Current methods for high throughput sequencing (HTS) for the first time offer the opportunity to investigate the entire ncRNAome in an essentially unbiased way. Here we present a web service called 'DARIO' that helps researchers to study short read sequencing libraries in a platform independent way and predict new ncRNAs. Small noncoding RNAs, including microRNAs, snoRNAs and tRNAs, represent a diverse collection of molecules with several important biological functions. Exploring small RNA biology or characterizing differential expression profiles by sequencing and comparing small RNA transcriptomes is also an exciting possibility to get more and deeper information about the world of non-coding RNAos (ncRNAs).


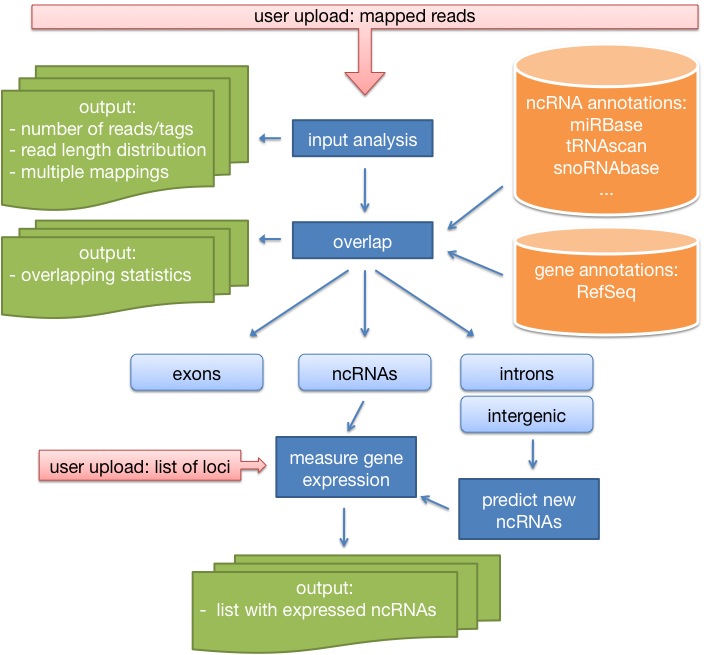
DARIO is a free web-server tool for the processing of small-RNAs data obtained from next generation sequencing techniques such as the Genome Analyzer of Illumina Inc. or Genome SequencerTM FLX (454 Life ScienceTM and Roche Applied Science). The input data are mapping positions of already mapped sequence reads stored in a bed-formatted file. Then, the tool performs several analysis steps and outputs detailed results. The analysis steps are the following:
- First analysis of the uploaded mapping file (Quality
control).
Here, the user gets a detailed analysis of the uploaded file (number of reads, number of tags, multiple mappings, read length distribution), as well as nice graphics showing where the reads mapped to (introns, exons, intergenic regions). This will help to evaluate the uploaded experiment. - Expression analysis of annotated ncRNAs.
In this step, the user will get a detailed analysis of the expression of already annotated genes. Since the expression values are normalized based on the number of mappable reads, the expression values are completely comparable with results of previous DARIO runs (differential expression). Furthermore, the user gets gets a link to the UCSC genome browser where he can take a look at the expression pattern. - Classification of previously unknown ncRNAs.
A set of new ncRNA candidates for miRNAs, snoRNAs and tRNAs is predicted. Furthermore, an overlap with well-known RNAz is computed to strengthen the result of real positives. The user gets an easy to use list with expression values of the new candidates, as well as the UCSC genome browser link, to see how the predictions look like. -
Expression analysis of user defined loci.
If the user decided to upload a set with new loci, these positions will be treated like ncRNA loci and a similar table with all the usefull informations will be generated. OF course, this table can also be downloaded.
DARIO requires mapped reads/tags and/or the user annoated loci stored in the bed format as input. Note that we only support data originating from experiments prepared with the short RNA-seq protocol and thus predominantly covers read lengths of about 22-24nt.
Mapped Reads/Tags
The mapped reads have to be in the following format:
#chr start end id nb of sequenced reads strand
chr1 20229 20366 GSM34290_325 50 +
chr1 1092347 1092441 GSM34290_326 3 +
User Annotations
If you want to upload your own annotation file or a file with some interesting locations, you have to create a bed file (filename must have correct suffix like .bed or bed.gz) with the following columns:
#chr start end id score strand type source
chr1 20229 20366 hsa-mir-1302-2 0 + miRNA miRBase_v16
chr1 1092347 1092441 hsa-mir-200b 0 + miRNA miRBase_v16
...
Mapping Tools and List of Supported Reference Genomes
As a first step, the sequenced reads must be mapped to a reference genome, using tools like:
DARIO supports the following assemblies as reference genomes:
Converting mapping results into BED file format
To convert the output of your read mapper to the bed format, you can use our map2bed.pl script. This script will not only create a bed formatted file, but also merge mapped reads to tags and try to pack the output file. This way, the file size will be minimized as much as possible. (Note: It might be necessary to explicitly select 'SAM format' as output format when running your mapping tool). The SAM file must contain header information.
SAM format (output from segemehl, Bowtie, BWA, ...):
perl map2bed.pl -i mapping.sam -f 1 -o upload.bed
SOAP format (output from SOAP):
perl map2bed.pl -i mapping.soap -f 2 -o upload.bed
Example BED files
Example BED-files containing previously mapped reads can be found on this page.
Converting mapping results into BAM file format
To convert the output of your read mapper to the BAM format, you can
use samtools.
Assure that the output format of your mapping tool
is the SAM format and call samtools as follows:
samtools view -bS yourFile.sam > yourFile.bam
NOTICE: You have to call samtools with the -S parameter, otherwise you will loose the header information of the SAM file, which is mandatory for our calculations!
You might want to inspect these hg18 example results. Also consider to take a look into the DARIO Tour (PDF) which gives detailed information about the DARIO usage and the output.
SUMMARY
In the summary section you will get informations like the start date/time of you job, how many entries your
bed-file has, the number of reads in you file (# of different ids) and the number of tags (# of unique ids).
QUALITY CONTROL
The quality control will give you a small summary of simple figures that will give you a first impression,
if and how good your experiment performed.
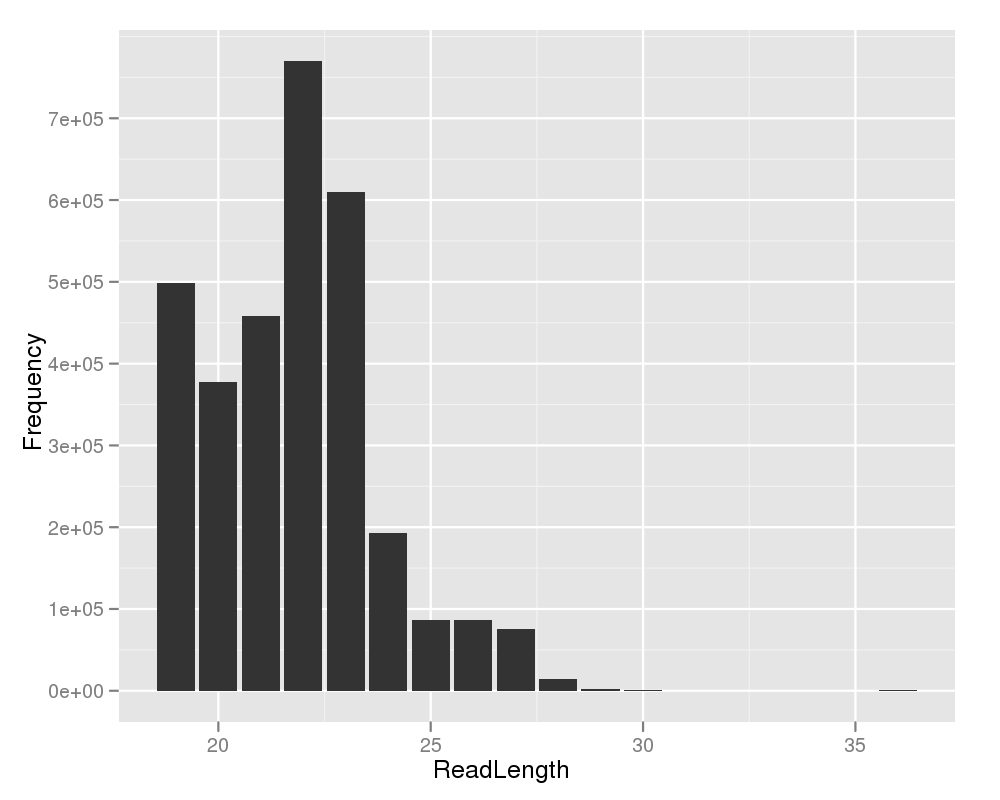
This plot shows you the read length on the x-axis and the amount of reads on the y-axis. Based on the read length distribution, the user can easily see, if the short read sequencing experiment performed properly. When using a short RNA preparation protocol, one would expect a peak at the length of 22. |
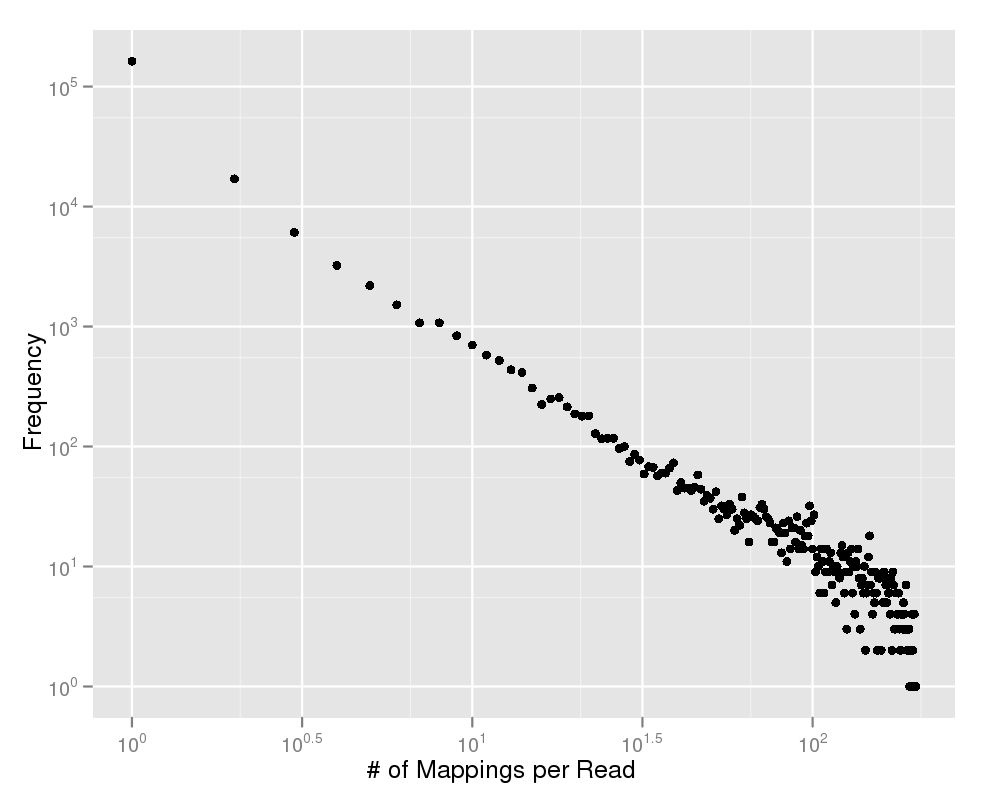
This plot shows you the number of multiple mappings on the y-axis and the amount of reads on the x-axis. Most of your reads should map to a unique position within your genome. If this plot gives any doubt to this statement, you should have a deeper look on the quality values of your experiment. |
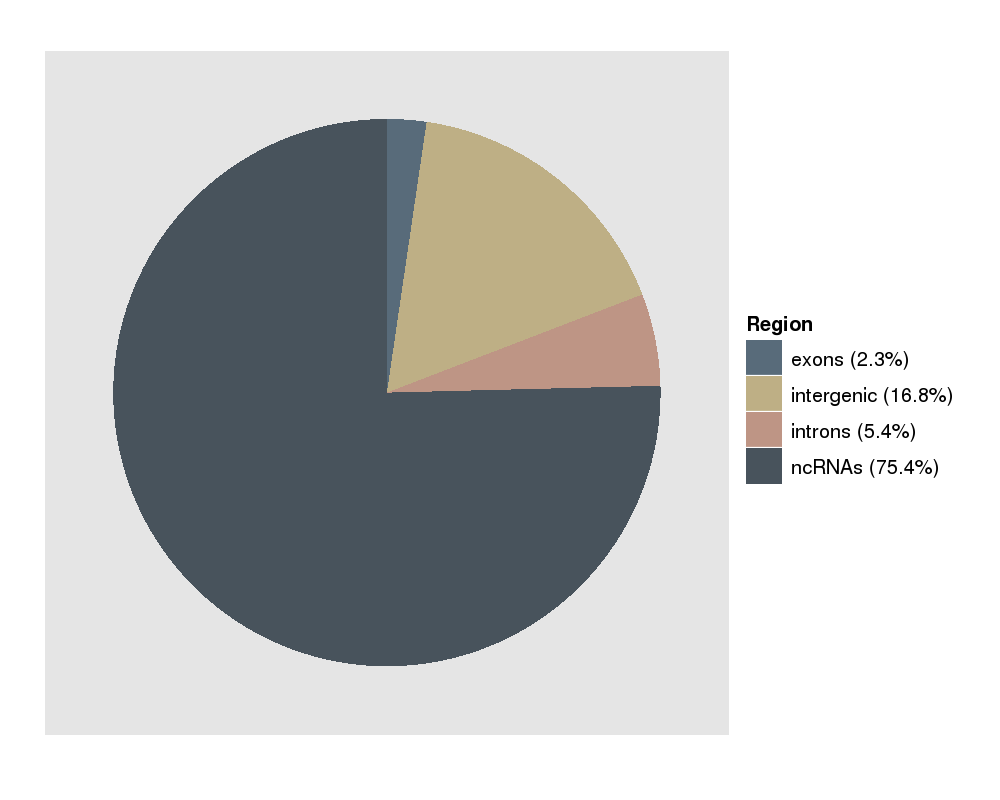
This plot shows you the fraction of reads that map to exons, introns, ncRNAs or not-annotated regions. The majority of the reads come from annotated ncRNAs. This picture is typical for a short RNA protocol. |
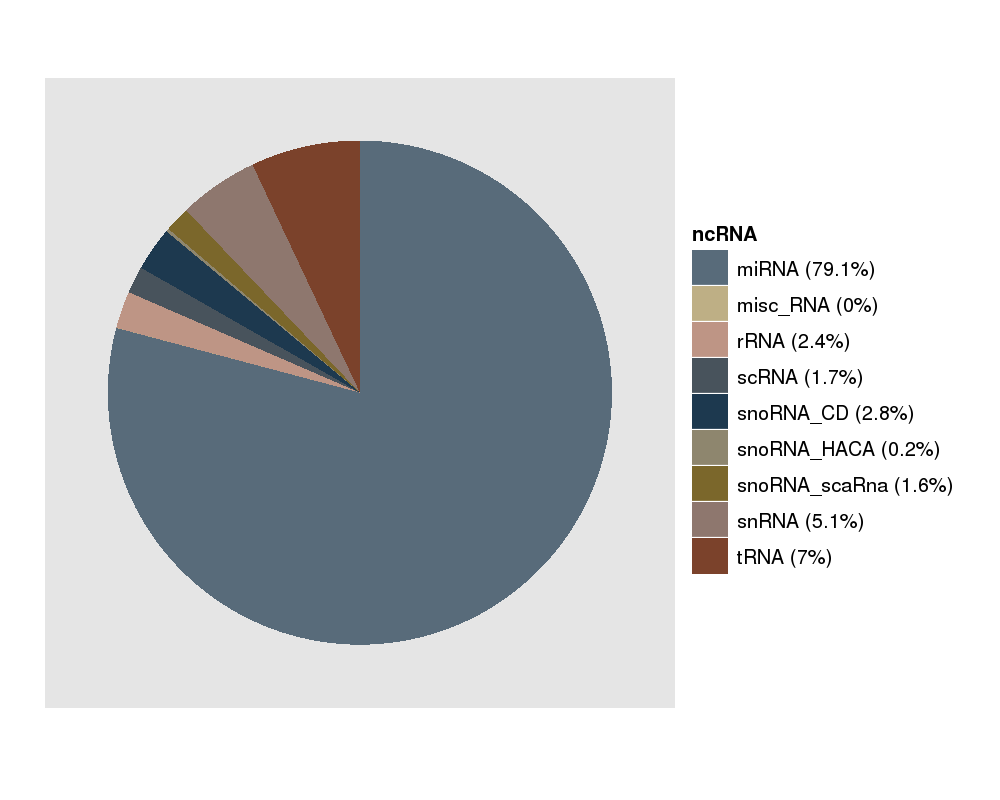
This picture gives you a feeling of the ncRNAs which are expressed in your experiment. miRNAs should be the majority here. |
ANALYSIS
In the main analysis reads are overlapped with annotated ncRNAs (miRBase, tRNAscan, snoRNAdb, etc.) and a list of expressed ncRNAs, itemized by ncRNA classes, is generated. You will find information like the number of reads mapping to annotated ncRNA loci and the normalized number of reads. To allow subsequent differential expression analysis, transcript measures are normalized based on their multiple mappings and the absolute number of mappable reads (RPM, Reads Per Million).
PREDICTION
For the prediction we use a previously published machine learning approach. Each class of ncRNA exhibits a chracteristic read pattern, which this method relies on to identify and classify new ncRNA candidates. For each candidate, a prediction score is given as well as a RNAz classification, if available. The tables of these candidates look similar to the expression tables of the annotated ncRNAs.
The DARIO Tour (PDF) also describes both analysis and prediction results.
Should I allow multiple mappings when mapping the reads to the reference genome?
Yes, we recommend to allow multiple mappings. Several microRNAs belong to microRNA families which have the same mature microRNA sequence. If you ignore these mature sequences in the mapping step, you might get misleading read patterns and 'expression' levels of these microRNAs. Furthermore, tRNAs are known to occur in multiple copies, sharing the same sequence and thus the same reads mapping to it. These multiple copies would be missed when only allowing uniquely mapped reads.
DARIO normalizes the read expression of reads mapping to multiple loci in the reference genome by dividing them by the number of mappings.
Will there be the possibility to upload raw reads to mapped automatically?
We are currently exploring the computational feasability of automatic mapping using our in-house aligner SEGEMEHL.
How to cite DARIO?
If you use DARIO for your research, please cite the following article:
DARIO: a ncRNA detection and analysis tool for next-generation sequencing experiments
Mario Fasold; David Langenberger; Hans Binder; Peter F. Stadler; Steve Hoffmann.
Nucleic Acids Research 2011; doi: 10.1093/nar/gkr357